


A very long time in the past, a very very long time in the past, designing supplies was a painstaking course of. For greater than 1,000 years, researchers have been making an attempt to create gold by mixing elements comparable to lead, mercury, and sulfur in simply the fitting proportions. Even well-known scientists like Tycho Brahe, Robert Boyle, and Isaac Newton tried their hand at what we name alchemy.
After all, supplies science has come a great distance. For the previous 150 years, researchers have used the periodic desk of parts to show us that totally different parts have totally different properties and that one ingredient can not magically rework into one other. Moreover, over the previous decade or so, machine studying instruments have enormously improved our capability to find out the construction and bodily properties of assorted molecules and supplies. New analysis by a bunch led by Zhu Li, a Tokyo Electrical Energy professor and professor of supplies science and engineering on the Massachusetts Institute of Know-how’s Faculty of Nuclear Engineering, guarantees a significant leap ahead within the capability to facilitate supplies design. Their findings are: December 2024 issue natural computational science.
Presently, most machine studying fashions used to characterize molecular programs are primarily based on density practical principle (DFT), a quantum mechanical method that determines the overall vitality of a molecule or crystal by analyzing the electron density distribution. Present. — That is principally the common variety of electrons positioned inside a unit quantity round every specific level in house close to the molecule. (Walter Cohn, who co-invented the idea 60 years in the past, received the Nobel Prize in Chemistry for it in 1998.) Though the strategy has been very profitable, there are some drawbacks, Lee stated. says. Uniformly fantastic. And secondly, it solely tells you one factor, the bottom whole vitality of the molecular system. ”
“Couple remedy” saves the day
His group now depends on one other computational chemistry methodology often called coupled cluster principle (CCSD(T)), additionally derived from quantum mechanics. “That is the gold customary in quantum chemistry,” Lee feedback. The outcomes of CCSD(T) calculations are way more correct than these obtained from DFT calculations and are as dependable because the outcomes at the moment obtained from experiments. The issue is that these calculations are very gradual to carry out on computer systems, he says. “And it is dangerous scaling: for those who double the variety of electrons within the system, the computation turns into 100 instances costlier.” Subsequently, CCSD(T) computations sometimes contain a small variety of about 10 electrons. It has been restricted to molecules with atoms. Something a lot past that merely takes an excessive amount of time.
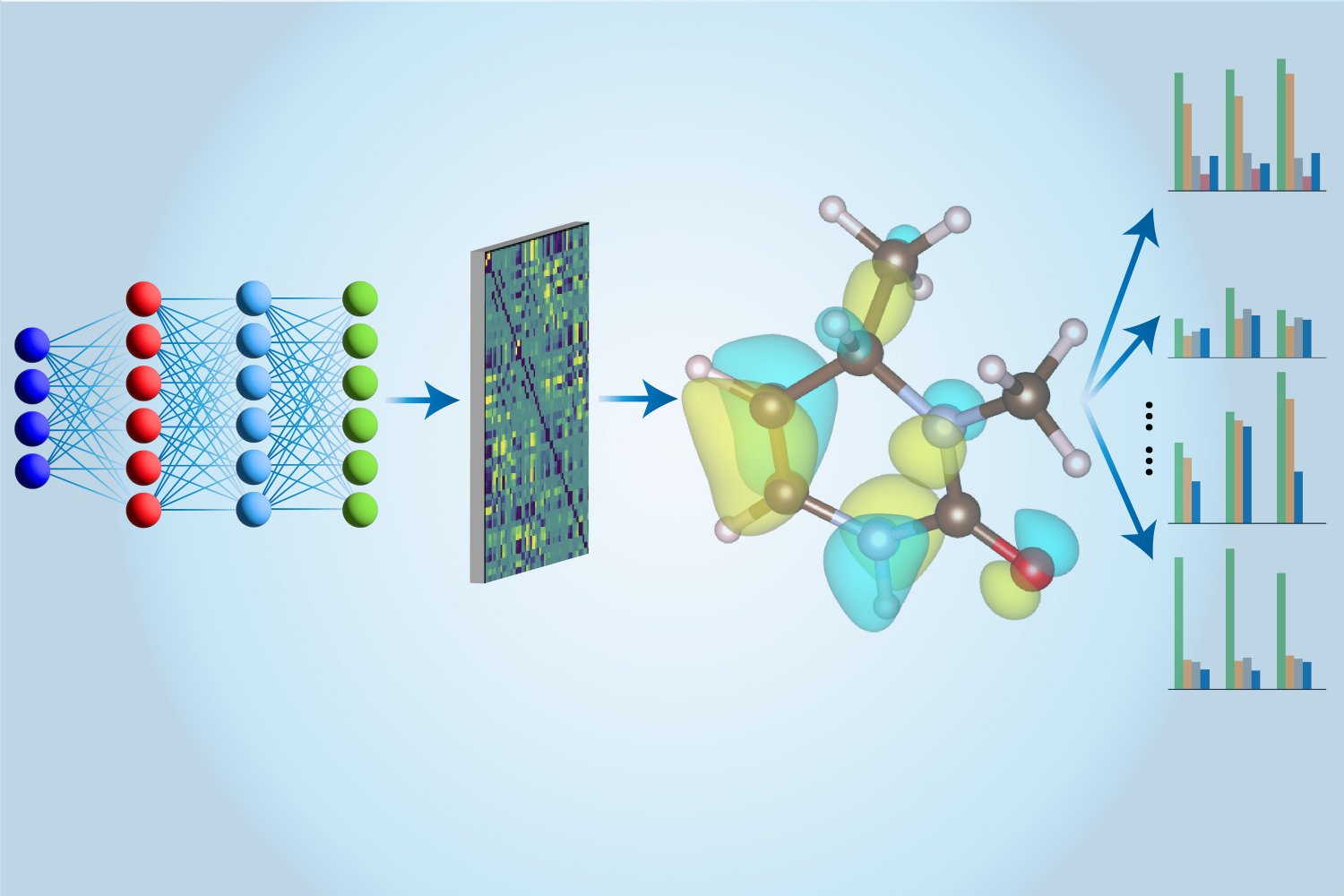
That is the place machine studying comes into play. The CCSD(T) calculations are first carried out on a traditional laptop, and the outcomes are used to coach a neural community on a brand new structure particularly devised by Lee and his colleagues. After coaching, neural networks can carry out the identical calculations a lot sooner by using approximation strategies. Furthermore, their neural community mannequin can extract extra details about molecules than simply their vitality. “In earlier analysis, individuals used a number of totally different fashions to guage totally different properties,” stated Hao Tang, a doctoral scholar on the Massachusetts Institute of Know-how, majoring in supplies science and engineering. says. “Right here we use just one mannequin to guage all these traits, which is why we name this a ‘multitasking’ method. ”
The “Multi-tasking Digital Hamiltonian Community” (MEHnet) has solved many digital properties, together with dipole and quadrupole moments, digital polarizability, and the photoexcitation hole (the quantity of vitality required to extract an electron from the Hamiltonian community). Masu. From the bottom state to the bottom excited state. “The excitation hole determines the frequencies of sunshine {that a} molecule can take in, so the excitation hole impacts the optical properties of the fabric,” Tan explains. One other benefit of their CCSD-trained mannequin is that it could possibly characterize not solely the bottom state but in addition the excited state. The mannequin may also predict the infrared absorption spectra of molecules, the place the vibrations of atoms throughout the molecule are related to vibrational properties that couple with one another, giving rise to totally different collective behaviors.
The energy of their method depends closely on community structure. Primarily based on analysis by MIT assistant professor Tess Smit“The group makes use of a so-called E(3) equivariant graph neural community, the place nodes characterize atoms and the perimeters connecting the nodes characterize bonds between atoms,” Tang stated. I’m. We additionally use custom-made algorithms that straight incorporate bodily rules associated to how molecular properties are calculated in quantum mechanics into our fashions. ”
check, 1, 2, 3
When examined with the evaluation of recognized hydrocarbon molecules, the mannequin of Li et al. It carried out higher than its DFT counterpart and was in shut settlement with experimental outcomes obtained from the printed literature.
Qiang Zhu, a supplies discovery knowledgeable on the College of North Carolina at Charlotte (who was not concerned within the examine), is impressed with the outcomes achieved thus far. “Their methodology allows efficient coaching on small datasets whereas attaining superior accuracy and computational effectivity in comparison with current fashions,” he says. “That is an thrilling examine that demonstrates the sturdy synergy between computational chemistry and deep studying, and offers contemporary concepts for growing extra correct and scalable digital construction strategies.”
The MIT-based group first utilized the mannequin to small nonmetallic parts (hydrogen, carbon, nitrogen, oxygen, fluorine, which might make natural compounds) after which to heavier parts (silicon, phosphorous, sulfur, chlorine and even platinum. After being educated on small molecules, the mannequin will be generalized to more and more bigger molecules. “Beforehand, most calculations concerned evaluation of a whole bunch of atoms by DFT and CCSD(T). We have been restricted to computationally analyzing only a few dozen atoms,” Li says. “Now we’re speaking about working with 1000’s and ultimately tens of 1000’s of atoms. .”
For now, researchers are nonetheless evaluating recognized molecules, however the mannequin might reveal never-before-seen options of molecules or hypothetical molecules made up of various kinds of molecules. can be utilized to foretell the properties of supplies. “The concept is to make use of theoretical instruments to select promising candidates that meet sure standards earlier than submitting them to experimentalists to examine,” Tan says.
It is all concerning the app
Zhu is optimistic about future potentialities. “This method has potential for high-throughput molecular screening,” he says. “This can be a activity the place attaining chemical precision is crucial to figuring out new molecules and supplies with fascinating properties.”
Demonstrating the flexibility to research giant molecules, maybe containing tens of 1000’s of atoms, “ought to enable us to invent new polymers and supplies” that may very well be utilized in drug design and semiconductor gadgets, Lee stated. Analysis into heavier transition metallic parts might result in the emergence of latest supplies for batteries, which are actually urgently wanted.
As Lee sees it, the longer term is extensive open. “It’s now not nearly one space,” he says. “Our final objective is to cowl the complete periodic desk with CCSD(T) stage accuracy at decrease computational value than DFT. It will enable us to resolve a variety of issues in chemistry, biology and supplies science. It is arduous to understand how extensive that vary is at this level.”
This analysis was supported by Honda R&D. Hao Tang acknowledges help from the Mathworks Engineering Fellowship. Among the calculations for this examine have been carried out on the Matlantis Quick Common Atomic Simulator, Texas Superior Computing Middle, MIT Supercloud, and Nationwide Power Analysis Scientific Computing.

{kind=link}